

Every cell talks: How Spatial transcriptomic tools are trying to decode the communication between them.
Omnis cellula e cellula

Dear community,
This will be slightly longer post so I kindly ask your patience as I navigate you through few interesting studies, I have also included a section trying to address the technical concepts in simple terms at the end of the newsletter (thank you for the feedback Ashwin). Also, If you like my posts and see scope for improvement in terms of understanding, leave me a comment and share amongst your friends. This also helps other readers on the platform discover the newsletter.
Everything in known existence co-exists, They have interactions either at an atomic level or at much more complex chemical levels. Whether it is gravity and time, or hydrogen and oxygen in a water molecule, Cells are no exception to this phenomenon. Uni-cellular organisms rely on chemical signals in their surrounding to navigate and find food, In many complex organisms including human beings cells rely on communication with their neighbours right from their early origins.
If you haven’t subscribed, click this yellow button!!!
Its free btw.,
As we continue in this journey of discovering spatial transcriptomics, we thereby stand at a crossroads of sorts. From imagining yourselves in the shoes of Robert hooke or Xavier Bichat or the ever-popular Ramon y Cajal; Across time and space we tried to do “thought travel” and over the centuries of this thought travel we have seen advances that would have not been imaginable. Cajal would have been amazed at the recent developments in neurology, for example, but not limited to being able to map neurons in the spinal cord of recovering mice from epidural electrical stimulation. Now, we stand at the foothills of emerging discoveries with the clouds surrounding the peaks like an inversion, except that you do not know the journey that is left to take to reach the summit to see a landscape of spatial biology. As we enter the second month of 2023, we have had a few more strides in spatial biology. One of the key advantages of leveraging spatial transcriptomics as discussed in Post 2 and Post 3 is that it’s slightly easier to identify cell niches and their interactions. In this context, two articles caught my eye over the last week.
1. Estimation of cell lineages in tumors from spatial transcriptomics data.
2. Screening cell-cell communication in spatial transcriptomics via collective optimal transport.
You might wonder, how are the papers useful together? So let’s find out through thought experiments. First, let me give you a breakdown of sorts about the papers individually, let’s briefly go through the key questions the authors tried to address, the innovative ways they have used and some takeaway points.
1. Estimation of cell lineages in tumors from spatial transcriptomics data.
Every cell that exists, especially in Eukaryotes originates from another cell. A yeast cell or an alga originates from its parent cell through multiple fission (in the case of algae) or through asexual reproduction by budding (in the case of brewers yeast). Imagine, if you are an enthusiastic home brewer; the yeast culture you have access to has a history and it might date back hundreds if not thousands of generations. Cell lineage denotes the development history, in your case, it would have been passed down from an abbey where the monks started brewing a few hundred years ago to your kitchen.
In the context of Humans, we can trace our cells back to the stages of the zygote from where sequential cell divisions and changes in the epigenome along with activation of several transcription factors determine which cell will belong to which organ. In living tissues, studying the lineages is essential to understand how cells interact with each other and the environment in which they reside. By understanding these relationships, researchers can gain insight into the fundamental mechanisms that control cell behaviour and the development of diseases. The attempts to identify the origin of a subset of the cells are often referred to as lineage tracing.
Before the advent of single-cell transcriptomics and spatial transcriptomics, lineage tracing was divided into two categories:
Prospective: Using markers such as GFP or ß-galactosidase. These are genes inserted along with the gene of interest so that they can be visualized in the tissue using microscopy.
Retrospective: Using markers that naturally occur and propagate along a cell lineage.
The era of spatial transcriptomics has set forward a less experimental approach ie., the reliance on prospective-only methods to estimate the cell lineages. With the availability of in-situ capturing strategies discussed in the other blog posts (check here and here); it is possible to capture the positions of the cells relative to their neighbours. However, there are challenges still; namely How to distinguish overlaps and mixed signals from multiple samples. Decomposition methods (One of which has been discussed here), are applied in characterizing the spatial and cellular landscape of tissues. This is particularly interesting in diseases such as cancer, where there might be multiple cell types in a small location.
The authors of the current study, I am writing about identified several shortcomings of the available decomposition methods. I have listed a few:
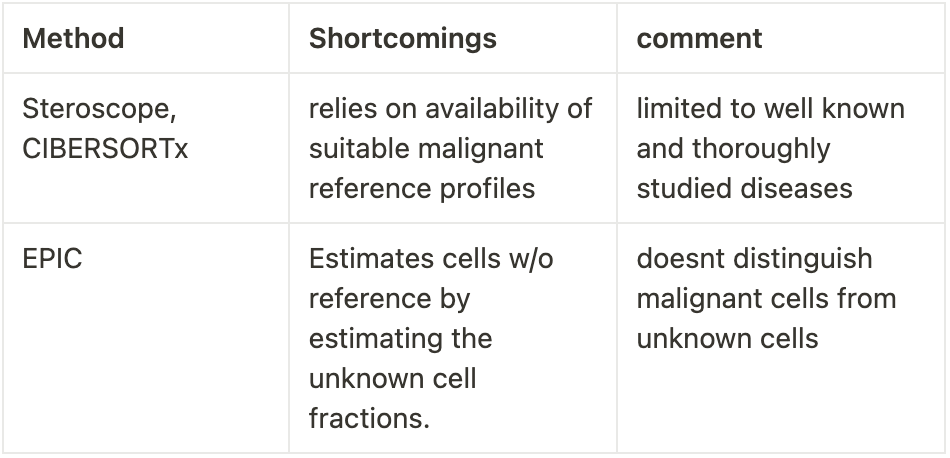
The authors suggest a new computational framework, SpaCET (Spatial Cellular Estimator for Tumors). The difference between SpaCET versus other existing methods is that It tries to address the challenges of tumour heterogeneity, tissue variation (like in invasive tumours), and immune cell integrity (remember that even immune cells try to attack the tumour, this is valuable information).
So how does this work,
Stage 1: Estimation of malignant cell fractions based on copy number alterations (CNAs) and malignant transcriptome signatures in common tumour types.
CNAs are events such as gene duplications that are common in most tumours, this leads to varied expression of proteins produced by these genes.
To accomplish stage 1, they created a gene pattern dictionary˚ of CNAs or tumour vs normal gene expression from 10k patients across 30 types of tumours.
The SpaCET algorithm searches malignant cell spots from spatial transcriptomics (ST) data and tries to match this dictionary.
Stage 2: Deconvolution of nonmalignant cell fraction and adjustment of cell densities using a unified linear model.
Using constrained linear regression*, SpaCET estimates cell lineages into immune lineages, stromal lineages, and unidentifiable components. SpaCET further deconvolves immune lineages into fractions based on prior knowledge of their parent lineages. The key point here is the hierarchical decomposition scheme will confine any result in the differences in sublineages from affecting the analysis of the parent cells.
Stage 3: Inference of intercellular interactions based on cell colocalization and ligand-receptor co-expression†.
They look at close contacts between cells within the same Spatial transcriptomic spot rather than between different spots because looking at two different spots may span several cells. The physical interactions allow them to further test the co-expression of ligand and receptor genes within the same spot.

Testing for performance:
For any new method proposed, a benchmark comparing the current method versus existing methods is a standard approach. These benchmark studies provide an insight into the performance of the methods, thereby convincing us the end users that there is some value to use these methods in our studies.
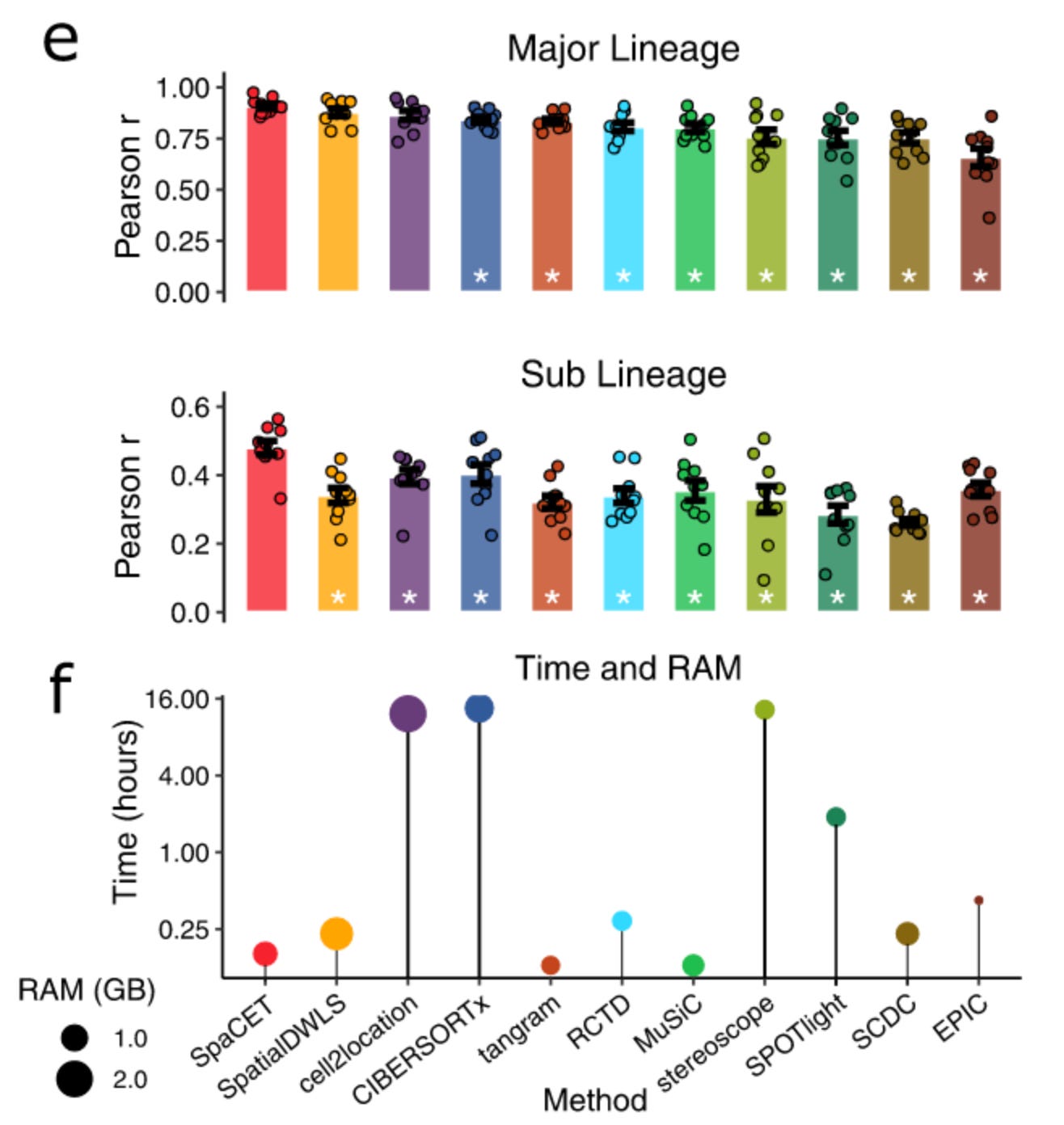
For the evaluation of SpaCET, the authors use a mix of simulated data; where the composition of the ST spot was known and real scSEQ data from various cell lineages from different tumour environments. Therefore, allowing them to compare the performance metrics. Simply put, they show that SpaCET was able to outperform other existing decomposition methods. They further use real tumour ST data along with H&E staining (these are often used in pathology labs to diagnose and observe tumours) to see if by only using SpaCET they can find different cell types identified by the pathologists as well.

Intracellular and ligand-receptor interactions.
Communication between tumour cells is an important mechanism by which the disease progresses. By using sc-SEQ datasets it is now possible to study the ligand-receptor co-expression across cell types; but due to the nature of the scSEQ experiment the locational proximity of the cells is usually lost, here is where the spatial sequencing helps us a lot.
SpaCET uses a two-step approach to identify intercellular interactions
Assessment of cell colocalizations: By investigating the cell contacts within the same spot of an ST experiment, they are able to calculate the correlation between cell-type pairs across ST spots based on estimated cell fractions. A strong correlation is likely to indicate the cell-type pair colocalization.
Since co-localization itself is usually not enough proof of physical interaction, They use Ligand-Receptor interactions within ST posts by computing an L-R network score for each spot.
The L-R network score indicates the likely intensity of ligand-receptor interactions at each location. By enriching these scores by leveraging scSEQ data in a breast tumour they show that SpaCET can accurately identify Cancer-associated fibroblasts and M2 macrophage interactions.
SpaCET is a framework for understanding the spatial organization
of cells in tumours and how spatial organizations influence cancer
progression. With the continuous accumulation of spatial transcriptomics data from clinical studies, we foresee that SpaCET provide mechanistic insights underlying many oncogenic processes and therapeutic solutions to bottlenecks of current antitumor treatments. Beibei Ru et. al,
2. Screening cell-cell communication in spatial transcriptomics via collective optimal transport.
On a similar note, Cang et al., recently published a new method to dissect cell-cell communication(CCC). The method COMMOT (COMMunication analysis by Optimal Transport) can be used to infer CCC in spatial transcriptomics data. These are a collection of algorithms that are developed to handle complex molecular interactions and infer spatial directionality and genes regulated by signalling using machine learning models. I have to agree, although I find this paper really interesting and the model useful; the language used here has been challenging to interpret.
While the previous method relied a bit on prior knowledge of receptor-ligand interactions and uses deconvolution of complex tissue; in their case cancer. COMMOT infers CCC by simultaneously considering numerous ligand-receptor pairs based on ST and scSEQ (spatially annotated) and summarizes and compares the directions of spatial signalling. It also helps to identify downstream effects (imagine, if you have a headache and consume paracetamol; the downstream effect of the drug is your pain relief) of CCC gene expression.
By using a method called Collective optimal transportª, a mathematical equation which is capable of preserving the comparability distributions and ensuring that the total signal in a region does not exceed the individual species amounts which enforce a certain range limit to signalling events and introducing an entropy regularizationº to enforce limits to the inequalities found for marginal distributions. They provide an elegant equation for a given spatial transcriptomic dataset with ligand species and receptor species; the collective optimal transport determines which of these species can be coupled and signalling strength from sender cell to receiver cell based on ligand and receptor. If interested in the equation please check the paper linked below.
So how does COMMOT work,
Here comes the technical jargon, I know I dread it as a biologist to see complex math, but let’s try to understand together.
Collective optimal transport:
Using non-probability mass distribution to control the marginals of the transport plan to maintain comparability between species.
Enforcement of spatial distance constraints CCC to avoid connecting cells that are far apart.
multi-species (here the species means different ligands and receptors) receptor and ligand to account for multi-species interactions.
Partial Differential equations:
They construct a PDE model to simulate CCC in space because direct validation of CCC for spatial data is difficult due to the lack of measurements of ligand and receptor proteins.
For each ligand-receptor pair and respective pair of cells, the CCC inference quantifies the ligand contributed by one spit to the ligand-receptor complex in another spot.
If cell A has ligand X and receptor Y; how much of the signalling sent out to Cell C somewhere close by actually receives the ligand/receptor? This is how we can interpret it in simpler terms. But I might be wrong here.
The directionality of the signal is then determined by interpolating the cell-by-cell CCC matrix to a vector feild˜ to identify the direction from which the signal is received or sent.

Based on this model, The researchers apply COMMOT to examine the development of the epidermis in human skin. They were able to identify four specific pathways that were known to have important roles in epidermal homeostasis. They also show that the signalling events in a 3D space move upwards. The key challenge for us is to remember that this relies on spatial transcriptomes and not direct interactions. So it is pretty cool that by using one method you are able to discern the signalling events in the skin.
They further make validation using different types of datasets such as MERFISH, STARmap, Slide-seqv2 and visium. This gives us the end users of the method to leverage this tool kit in our studies to understand the signalling events we are interested in our experiments.
One of the most thoroughly studied signalling events in biology belongs to the drosophila embryonic signalling ˚˚. The authors show that by using COMMOT they are able to determine the signalling events that occur in drosophila embryos. They also were able to give examples of identified positively, negatively, and partially differentially expressed genes associated with Dpp signalling.


A brief summary
We looked into two methods that are unique in their approaches but try to solve a similar problem. The problem of which cell talks and where is the information received. In the grand scheme of things, It is my opinion that by using a single experimental data set and being able to leverage several tools to understand biological phenomena better we would be in a position to delineate the complexities in biology. Here, SpaCET has been applied for tumours and COMMOT has been applied for signalling. Both teams are inherently interested in cell communication, but their approaches to the problem are different from each other. While exploring options to use tools in our data, we have to see what is our hypothesis and how effectively a tool can address the challenges we face in our experimental hypothesis.
References
Estimation of cell lineages in tumors from spatial transcriptomics data by Ru et al.
Lab socials: Twitter, Lab, Software package
2. Screening cell-cell communication in spatial transcriptomics via collective optimal transport.
Lab Socials: Lab, Software package
Concepts briefly explained:
˚Gene pattern dictionaries are collections of gene expression patterns collected from different types of cells, tissues, and diseases. The patterns are used to identify the type of cell or tissue that is present in a sample. The gene expression patterns are determined by analyzing the expression of specific genes in the sample. This information can be used to identify the cell type or tissue that is present in the sample. Gene pattern dictionaries can also be used to identify genes that are associated with a particular disease, providing insight into the underlying cause of the disease.
*Constrained linear regression is a type of linear regression where certain constraints are applied to the model. These constraints can limit the value of the regression coefficients, or they can restrict the range of values that the dependent variable can take. This helps reduce overfitting and improves the accuracy of the model. Constrained linear regression can also be used to identify outliers and reduce the effects of multicollinearity.
†Ligand-receptor co-expression is the simultaneous expression of a ligand (a molecule that binds to another molecule) and its receptor (a molecule that binds to a ligand) on the same cell surface. This type of co-expression allows for communication between the two cells and is a key component of many biological processes, such as cell-cell signalling, cell migration, and cell adhesion.
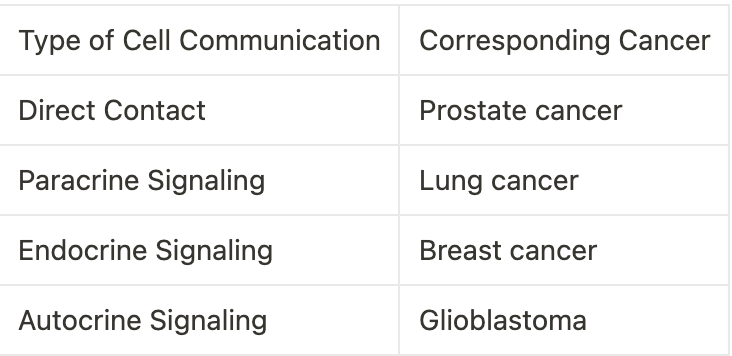
Communication between Cancer Cells.
Cancer cells can communicate with one another through a variety of methods, including direct contact, paracrine signalling, endocrine signalling, and autocrine signalling. This type of communication is essential for cancer cells to survive, grow, and spread. The following table outlines the various types of cell communication and their corresponding cancers:
ˆA double-blinded study is a clinical research study in which neither the participants nor the researchers are aware of which participants are receiving the treatment or the control. This type of study is designed to reduce bias and ensure that the results are purely based on the effect of the treatment and not on any external factors. Double blinding is often used in randomized controlled trials, which are considered the gold standard in clinical research.
ª Collective optimal transport is a mathematical optimization problem that is used to find the most efficient way of transporting a group of items from one place to another. It is based on an equation that considers the distances between the items, the costs associated with transporting them, and any constraints that may be imposed on the transport. The goal of the equation is to find the most cost-effective way of transporting the items while respecting the given constraints. The equation works by finding the path that minimizes the total cost of transport, taking into account the distances, costs, and constraints. This can be used to find the most efficient way to transport goods, people, or even data.
º Entropy regularization is a technique used to improve the accuracy of a machine learning model. It works by adding a penalty term to the model that penalizes the model for making predictions that are too confident. This penalty encourages the model to make more conservative predictions, which can lead to more accurate results. Entropy regularization can be used in a variety of machine learning algorithms, including regression, classification, and clustering. In simple terms, entropy regularization works by discouraging the model from making overly confident predictions and encouraging it to make more accurate predictions.
˚˚The Dpp and Wg signalling pathways are key players in the development of the Drosophila embryo. Dpp signalling is necessary for the formation of the dorsal-ventral (DV) and anterior-posterior (AP) axes in the embryo, while Wg signalling is necessary for the formation of the lateral (L) and proximal-distal (PD) axes.
The Dpp signalling pathway begins with the secretion of a Dpp ligand from cells in the dorsal region of the embryo. This ligand binds to its receptor, the Thickveins (Tkv) receptor, on cells in the ventral region of the embryo. This binding leads to the activation of a signal transduction cascade, which results in the phosphorylation of the transcription factor Mothers Against Dpp (Mad). The phosphorylated Mad then binds to the DNA and activates the transcription of various target genes, which are necessary for the formation of the DV and AP axes.
The Wg signalling pathway begins with the secretion of the Wg ligand from cells in the lateral region of the embryo. This ligand binds to its receptor, the Wingless (Wg) receptor, on cells in the proximal-distal region of the embryo. This binding leads to the activation of a signal transduction cascade, which results in the activation of the transcription factor Dachsous (Ds). Ds then binds to the DNA and activates the transcription of various target genes, which are necessary for the formation of the L and PD axes.
The Dpp and Wg signalling pathways are essential for the proper development of the Drosophila embryo. They initiate and proceed through the secretion of their respective ligands, which bind to their respective receptors. This binding leads to the activation of signal transduction cascades, which results in the activation of transcription factors and the transcription of target genes. These target genes are necessary for the formation of the four main axes of the embryo.

{kind=link}
Keep me Caffeinated by scanning the QR code and donating what you like.

I have found a short form summary of Optimal transport published in nature methods:
https://www.nature.com/articles/s41592-022-01729-3