

Peering through the tissues: Roots of innovation #3
An hybridization approach for spatial transcriptomics and application

Dear Members of the community,
A quote I came across not long ago in many parallels is profound and striking for us, irrespective of what field of research or career you are pursuing; It is often one’s curiosity that sets forward a path in life that you are likely taking. The quote is as follows
“Physicists are made of atoms. a physicist is an attempt by an atom to understand itself” - Michio Kaku, Parallel worlds: A journey through creation, Higher Dimensions and the Future of the Cosmos
Similarly, for me;
Biologists are made of cells. A biologist is an attempt by the cells to understand themselves.
The beginnings of my journey into biology, peering through a microscope to see the cellular organization of onion epidermal cells in a dusty lab at a school in Chennai when I was probably in the 6th grade; My journey might have been set by that one observation and a cumulative set of events that led to me pursuing the current field of study. I don’t often reflect on this, I guess it is a part of modern-day adulting not to dwell and reminisce and how it has influenced my present.
So, Harsha why are you being philosophical and talking about the past and the present you ask?
In my view, most if not all modern technologies have been enabled by incremental improvements in various fields. The modern sequencing machine such as the widely used Illumina novaseq would not be possible without the advances in microfluidics, semiconductors, and imaging. So is the case for current advances in spatial transcriptomics. I will take this opportunity to make a case for incremental improvements over decades that led to current bleeding-edge technology.
In 1994, Long before I knew there existed a field called molecular biology or biology for the matter of fact; Mats Nilsson published a paper on padlock probes (PLP) during his PhD studies at the then Beijer Laboratory of the department of medical genetics at Uppsala university (This may have been renamed to the department of Immunology, Genetics and Pathology now). As the authors mention, the challenge was identifying single-copy gene sequences amongst sequence variants. To this end, they developed oligonucleotide probe molecules composed of two target-complementary segments connected by a linker that may be used for detection. This “padlock” probe functions via hybridization of the two segments to a target sequence (imagine a small strand of DNA is the body of the lock and two segments represents the loop of the lock); This hybridization allows the probe segments to be covalently joined by the action of a DNA ligase. Two things are key here;
Simultaneous hybridization of the two segments provides higher specificity.
Ligation allows the distinction of non-target sequences because the mismatch won’t allow effective DNA amplification.
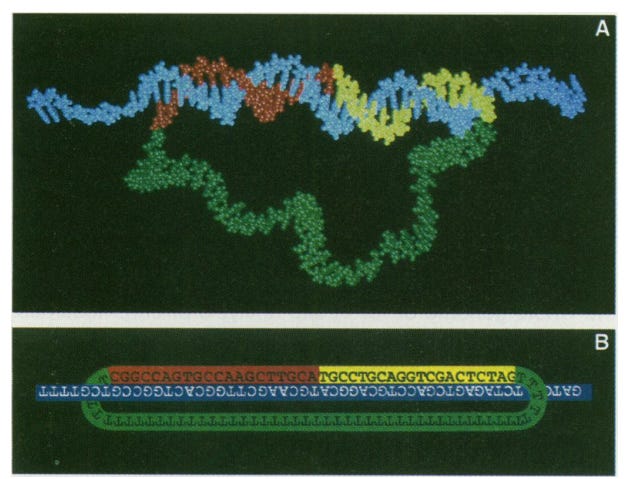
This technological advance, in retrospect, seems to be trivial in the current context. But, I am sure at that period when there was not a lot of modern technology and what I would call the beginning of the “large-scale sequencing era” the ability to specifically target a short segment of DNA and then with greater specificity detect mutations in DNA is a quantum leap for that time.
This was the early beginning of the more modern spatial sequencing methods such as ISH. Another advancement I want to bring to your attention is the Proximity Ligation assay a further advancement of Padlock probes which allowed the detection of proteins and other macromolecules within a cell. Briefly, PLA relied on tagging two antibodies (they recognize the macromolecule of interest) with oligos which are hybridized by ligation and a Roll Circle Amplification (RCA) is performed to give us a readout.
What do pad-lock probes and hybridization-based spatial sequencing methods have in common?

In 2020, The article of interest for the current theme was published in a study steered by Mats Nillson with the primary author Daniel Gyllborg in the journal Nucleic acids research. The authors describe an iteration of In situ sequencing named HybISS, or Hybridization-based in situ sequencing.
If you are curious about what category this approach falls under check out Part 1 of the series.
Essentially, they modified the probe design allowing for a new barcoding system via sequence-by-hybridization chemistry. It allows for HybISS technology to work as a targeted amplification detection method for improved spatial transcriptomic visualization.
What’s new in the approach:
It implements combinatorial barcoding, allowing for more robust detection of molecules.
Allows for an upscaled spatially resolved gene expression.
Supports for the accommodation of larger gene panels; This was a significant limitation in Sequence-by-ligation methods such as ISS.
How does it work:
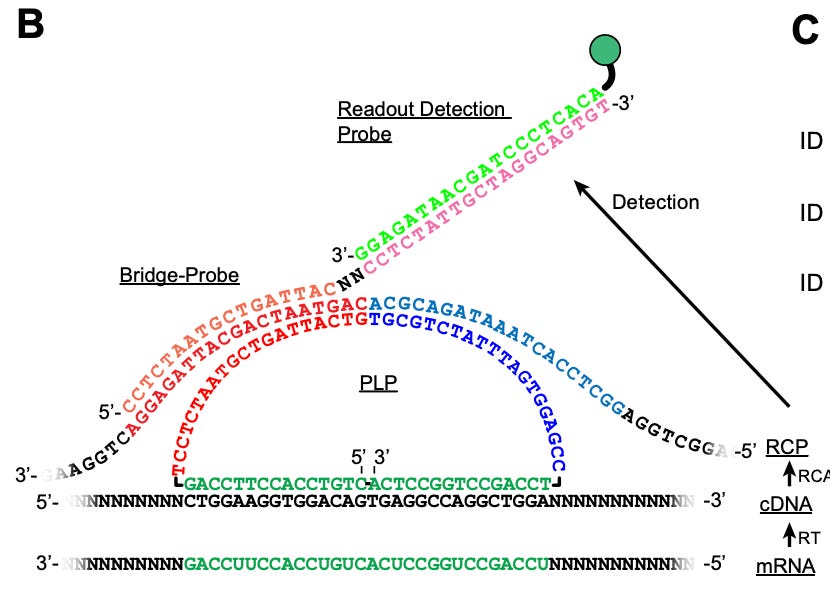
PLPs contain two parts (the structure of PLP is shown in the image above):
Unique ID sequence that “targets“ gene of interest.
Anchor sequence shared by all PLPs.
The tissues are fixed and permeabilized followed by reverse transcription with random primers. This is followed by fixing the tissues to arrest RT reactions.
A hybridization step is performed using PLPs.
RCA is then performed using phi29 polymerase. And a quenching step is performed to reduce noise
Bridge probes are then hybridized. These probes act as an intermediate to the readout detection probes.

HybISS method overview: A) Overview of the ISS style of steps (HybISS varies in the implementation of the probe design). B) HybISS: Each cycle combines the bridge probes to the RCP and reads them out with fluorophore-conjugated readout detection probes. consecutive stripping and rehybridizing allow for the detection of the transcripts. Figure from Gyllborg et al., In the paper, the authors go on to show how HybISS can be applied on a mouse coronal brain section using PLPs to map 119 genes over 5 cycles in the section. They show that using HyBISS the expression distribution of individual genes was possible within the mouse visual cortex, hippocampus and thalamus. Why is this important: Well the method is generally applicable and robust enough to be used across tissues.

As you can see, an older technological advancement in the 1990s has indeed laid the ground for a much newer method of spatial transcriptomics. As mentioned in blog posts (Take a look if you missed them; links below) convergence of various methods and overall incremental updates unlock new avenues in understanding tissue architecture in healthy and disease conditions. Understanding these architectures is crucial for biologists and bioinformaticians to evaluate the alterations in disease conditions, thereby contributing to the potential development of drugs against complex diseases.

Direct RNA targeted in situ sequencing: The next iteration.
In the current post, we have seen an example of a convergence i.e., utilization of PLPs in a spatial transcriptomic context. But, there are more challenges that need to be solved, i.e., there is always room for improvement in any method. In a subsequent paper by Hower Lee from Mats’s lab they experimented with the following question
Since ISS suffers from low transcript detection efficiency, can we circumvent the reverse transcription step to increase detection efficiency?
The reverse transcription step essentially converts mRNA molecules to cDNA to be probed by consequent spatial transcription experiments. This also leads to inefficiencies along with PLP hybridization and ligation. Remember, any additional step in an experiment introduces biases and interference with final signals; So by skipping one it can be assumed that one can increase the signal by a certain magnitude.
By using CARTANA’s (now 10x Genomics) direct RNA (dRNA) probing chemistry; which retains the fundamental benefits of cDNA-based ISS technology, They compare and contrast the two methods to see which has a better advantage in terms of transcript resolution. Similar to the PLP in HybISS, they designed oligos with a 20 nt long unique ID assigned to the gene of interest only this time the binging is targetted against the mRNA instead of cDNA.
For the comparison of dRNA-HybISS to cDNA-HybISS they selected four genes marking different cell types in the mouse brain coronal section:
Cd24a: Ependymal cells
Mbp: Oligodendrocytes
Lamp5 and Slc17a7: Excitatory neurons
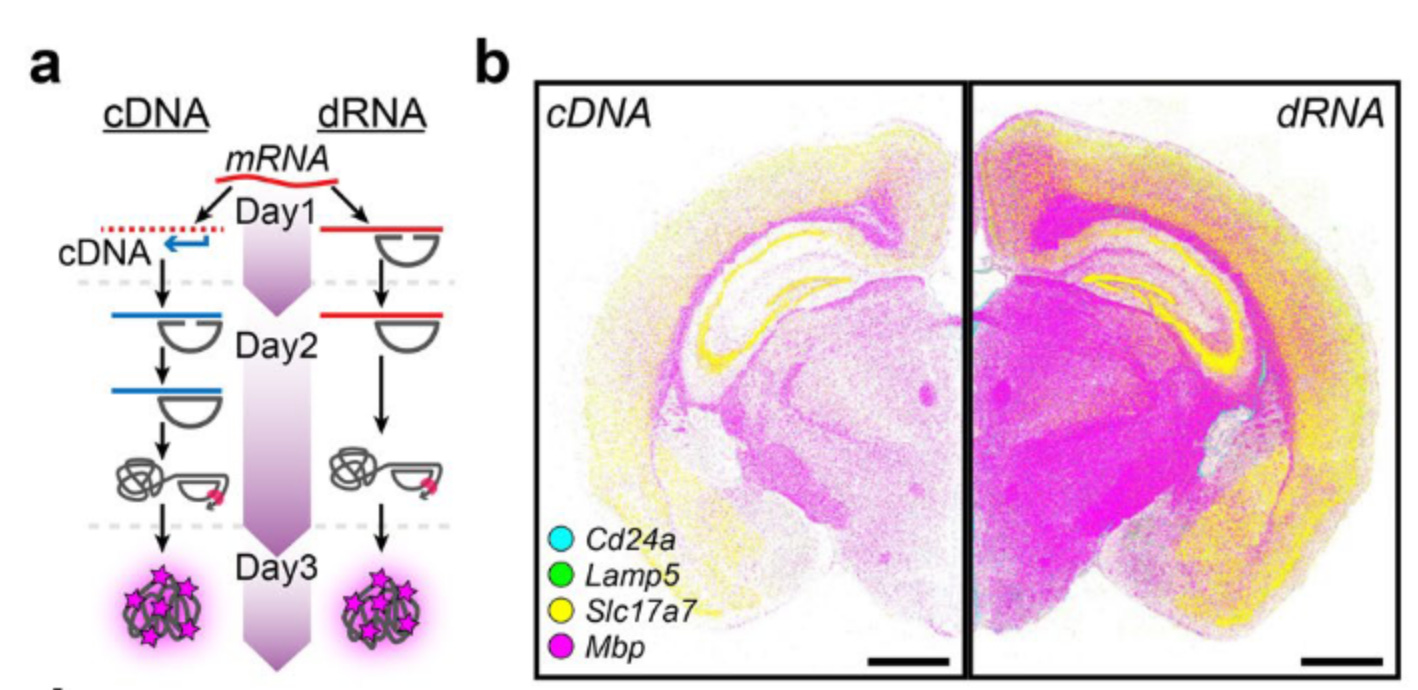
Using a dRNA-HybISS method they were able to show:
Increased detection efficiency in comparable images of Regions of Interest.
A five-fold increase in detection efficiency
comparable signal-to-noise ratio between the two approaches.
I implore you to take a look at the paper to further understand the method.
Overview of Analysis pipelines.
As a bioinformatician, you might be interested in the tools available for spatial transcriptomic data analysis. I have compiled a list of four tools to get you started along with a brief overview.
STARFISH: a toolkit for the analysis of large-scale, spatially-resolved transcriptomics data, including spatial transcriptomics data. The workflow in STARFISH typically involves processing the raw imaging data to extract gene expression information, normalizing the data, and performing clustering and visualization of the results. STARFISH also provides tools for cell type identification and spatial resolution improvement, which are important for the interpretation of spatial transcriptomics data.
SEURAT: Seurat is a popular R package for single-cell data analysis, including spatial transcriptomics data. The workflow in Seurat typically involves quality control and normalization of the data, dimensionality reduction using techniques such as Principal Component Analysis (PCA), clustering of cells based on gene expression profiles, and visualization of results through heatmaps, t-SNE plots, and spatial maps.
ILASTIK: Is designed to provide an intuitive and interactive interface for users, making it accessible to a wide range of researchers, regardless of their computational expertise. The workflow in Ilastik typically involves processing the raw imaging data to extract gene expression information, normalizing the data, and performing clustering and visualization of the results. Ilastik also provides machine learning algorithms for cell type identification and spatial resolution improvement, which are important for the interpretation of spatial transcriptomics data.
Ilastik has gained popularity in the field of spatial transcriptomics due to its user-friendly interface and its ability to handle large datasets. It also has a range of built-in algorithms for image analysis, including image segmentation, cell type identification, and spatial resolution improvement, making it a versatile tool for spatial transcriptomics analysis.
DeepCell: an open-source tool for deep learning-based image analysis, including the analysis of spatial transcriptomics data. It is designed to provide an end-to-end workflow for the analysis of spatially-resolved transcriptomics data, from data pre-processing to visualization and interpretation of results.
The workflow in DeepCell typically involves data pre-processing, including image normalization and gene expression quantification, followed by the training of deep learning models for cell type identification, spatial resolution improvement, and clustering. The results are then visualized using techniques such as heatmaps, t-SNE plots, and spatial maps.
Summarizing the evolution
Padlock probes have been crucial to the growth of spatial transcriptomics, as they enable the precise mapping of RNA molecules to specific locations in cells and tissues. Spatial transcriptomics, in turn, requires continuous advancements in various fields, such as microscopy, sequencing, and histochemistry, to produce more accurate and comprehensive data on gene expression patterns in space and time. These incremental innovations have enabled researchers to understand biological processes and disease mechanisms deeper, leading to new therapeutic targets and diagnostic approaches.
The integration of spatial transcriptomics with other high-throughput technologies such as single-cell sequencing and imaging mass cytometry has allowed researchers to study gene expression and cellular function in unprecedented detail. This has provided new insights into complex biological processes, such as tissue development, immune response, and disease progression. The results from these studies have broad implications for our understanding of human health and disease, and the development of new diagnostic and therapeutic approaches.
In addition, the development of padlock probes and spatial transcriptomics has also opened up new avenues for interdisciplinary research, where scientists from different fields, such as physics, engineering, and computer science, can collaborate to solve complex biological questions. This interdisciplinary approach is critical for advancing our knowledge of complex biological systems and for developing new technologies to study them.
Overall, the contributions of padlock probes and spatial transcriptomics to the fields of biology and medicine cannot be overstated. These powerful tools have changed the way we think about gene expression and cellular organization and have paved the way for many exciting new discoveries in the years to come.
A few buttons to click; spread the knowledge and shine the light of wisdom.
You can reach to me and share your thoughts here at any point in time

References and further reading
Padlock probes: circularizing oligonucleotides for localized DNA detection by Mats Nilsson and co-authors. The article is behind a paywall, drop me a message in the subscriber chat or in the comments for a copy of the report.
Thank you for your valuable time; I recognize its importance to all the community members.
By sharing knowledge with you my community members, I am able to learn so much in this journey along with you. The below is completely optional but if you like to donate a cup of coffee; I appreciate it very much.
scan the QR below or click here :)
