

When Algorithms Meet Biology: A Deep Dive into Biomedical Deep Learning
"For the very first time in human history, biology has the opportunity to be engineering, not science." Jensen Huang

Dear Valued Community Members,
It has been quite a while since my last post, I had to put the tasks on hold due to a set of unforeseen events with my wife’s health. She had been diagnosed with an adenocarcinoma, and she needed my complete support during her treatment and recovery period. I hope my unexpected hiatus will slowly halt.It took some mental toll on me as well; restarting this newsletter was a difficult enterprise during the last year or so. However, I will try my best; let’s see how this journey restarts and continues.
Without further ado, Let’s start with the story
PS: Please leave comments so I know I am not shouting into the void!
There's a unique thrill in exploring life's intricate machinery. Biology, with its dynamic systems and complex interactions, has always presented a fascinating frontier. We are now in an exciting era. The sheer volume of biological data—from genomic blueprints to protein structures, immune responses, and drug interactions—calls for a new kind of explorer. Deep learning is emerging as an indispensable tool for many of us navigating this vast information ocean. It's more than a buzzword; it's becoming a vital compass. This technology helps us chart unknown cellular territories and even predict how life responds and adapts.
Why Deep Learning is Becoming Biology's New Microscope (and Crystal Ball*)
Consider our traditional methods for understanding biology. Rule-based systems and classical statistics have their merits, but they often struggle with life's sheer scale and non-linear nature. Deep learning models, however, possess a remarkable ability to learn from raw data. They can discern patterns in sequences, structures, or images without explicit pre-programming. This capability is a true game-changer.
Automated Sleuthing: These models identify critical features in complex datasets, much like a skilled detective finds overlooked clues.
Handling the Haystack: They can analyze terabytes of multi-omics data, efficiently finding valuable insights.
Learning and Adapting: Techniques like transfer learning and self-supervision make these AI systems versatile. They can apply lessons from one biological puzzle to solve others.
In this post, I'm excited to guide you through three pioneering studies. These studies demonstrate how deep learning is actively transforming biomedical research, not just supplementing it. We will explore:
Cancer's Hide-and-Seek: How AI predicts when protein mutations lead to mislocalization—a common tactic cancer uses to develop.
Decoding the Immune Handshake: A novel model that examines how immune cells recognize threats, a critical step for better vaccines and cancer treatments.
Speeding Up the Hunt for RNA Drugs: Using deep graph learning to rapidly screen for potential RNA-targeting medicines, a newer area in drug discovery.
Join me as we dissect the science, the clever algorithms, and the real-world impact of these developments. As always, I welcome your thoughts and questions in the comments!
Section 1: When Proteins Go Astray – Deep Learning Pinpoints Cancer-Causing Mislocalization
The Biological Conundrum: Why a Protein's Address Matters Life and Death
Imagine a city where every worker has a specific zone. Proteins are the cell's workforce. Their correct locations—the nucleus, cytoplasm, and so on—are vital for their function. Small "address labels" in their sequences, known as nuclear localization signals (NLS) or nuclear export signals (NES), guide them. What happens if these labels are altered by a genetic mutation? The protein can end up in the wrong place. This misplacement can prevent it from doing its job or, worse, cause cellular chaos. Such mislocalization is a known issue in many diseases. It is a particularly insidious strategy in cancer. For example, if tumor suppressor proteins cannot reach the nucleus, or if oncogenic proteins are trapped where they cause maximum damage, the results can be severe.
Historically, identifying these "shuttling-attacking mutations" (SAMs) has been a slow and often fragmented process. Most predictive tools relied on known NLS/NES motifs. But many proteins use unconventional routes or signals, posing a challenge for these tools.

Enter PSAM: An AI Detective for Protein Shuttling
To address this, researchers developed PSAM (Prediction of Shuttling-Attacking Mutations). PSAM is a deep learning model designed to predict, from sequence alone, if a mutation will likely misroute a protein. Its strength lies in its ab initio approach. PSAM doesn't just search for known address labels; it learns the complex sequence "language" that governs nuclear localization.
Peeking Under the Hood: How PSAM Works
PSAM uses a sophisticated hybrid architecture, like a team of specialists:
Convolutional Neural Networks (CNNs): These detect local patterns or motifs in the protein sequence important for its transport.
Bidirectional Long Short-Term Memory (BiLSTM) networks: These networks "remember" long-range dependencies. They understand how distant amino acids in the linear sequence might collectively affect localization.
Attention Mechanisms: This clever component allows the model to weigh the importance of different sequence parts in its prediction. It effectively highlights critical residues.
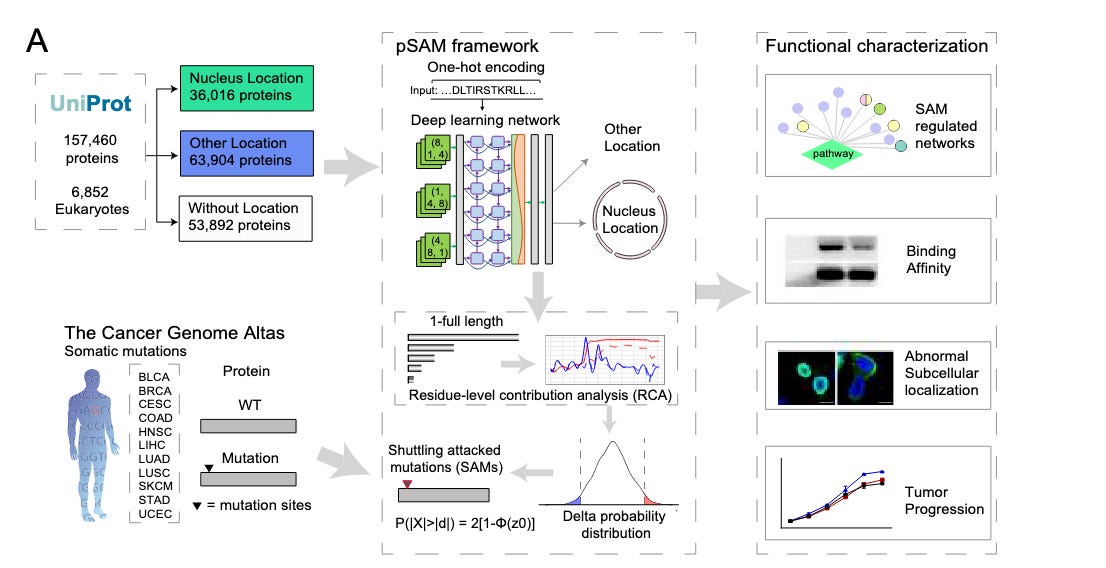
Model Workflow
[Protein Sequence Input (Wild-type or Mutated)]
│
▼
[One-Hot Encoding & Embedding (Numerical conversation)]
│
▼
[CNN Layers(Feature extraction)]
│
▼
[BiLSTM Network (Contextual understanding)]
│
▼
[Attention Mechanism & Residue-Level Contribution Analysis (RCA) (Pinpointing key sites)]
│
▼
[Output: Nuclear Localization Probability & Predicted Mutation Impact]
Training and Putting PSAM to the Test
PSAM was trained on a large dataset of eukaryotic protein sequences with known localizations from UniProt. Its performance was thoroughly evaluated:
High Accuracy: PSAM showed top-tier performance in predicting nuclear localization. It outperformed existing tools, achieving impressive AUC values (often above 0.98) on test datasets.
Decoding Determinants: The Residue-level Contribution Analysis (RCA) offers interpretability. It helps identify specific regions (Determinants of Nuclear Localization or DNLs) that PSAM finds important. Many DNLs are at the N-terminus, a common NLS location, but they can appear elsewhere, showing PSAM's ability to find unconventional signals.
Validating SAMs: Researchers analyzed mutations across 11 cancer types from The Cancer Genome Atlas (TCGA). SAMs constituted about 6.7% of mutations in known NLSs. Mutations in these DNL regions were more frequent and more damaging.
Experimental Confirmation: Predictions were confirmed experimentally. The team validated a dozen SAMs. For instance, mutations like R14M in PTEN and P255L in CHFR disrupted nuclear localization by affecting binding to importin proteins. These mislocalizations then altered downstream signaling pathways, neutralizing the proteins' tumor-suppressive functions. Immunofluorescence staining clearly showed altered localization for several predicted SAMs, like KAT8 p.R144C.


My Perspective:
PSAM's move beyond simple motif-matching is genuinely exciting. The ab initio learning, with RCA for interpretability, marks a significant advance. We can now start uncovering novel regulatory sequences for protein trafficking previously unknown to us. The finding that DNLs often overlap with DNA-binding domains also raises intriguing questions about the dual roles of these regions.
The authors acknowledge challenges. Current NLS/NES annotations are incomplete. Defining a "negative dataset" is also complex; a protein not observed in the nucleus might still go there under specific conditions. However, PSAM offers a potent new analytical lens. Systematically analyzing cancer mutations for their shuttling-attacking potential is a major contribution. It adds a new layer of functional annotation for cancer genome variants, helping prioritize mutations that drive disease by disrupting this key cellular process.
Future work integrating post-translational modifications or environmental cues could make such models even stronger. The public iNuLoC database and webserver they built are also vital for progress, embodying the scientific spirit of sharing tools and data.
Section 2: The Immune System's Secret Handshake – AI Deciphers Antigen Binding
The Biological Puzzle: Immunity's Double-Check System
Our immune system operates with remarkable precision. A core function is distinguishing "self" from "non-self," like invading pathogens or cancerous cells. Two critical interactions, or "handshakes," determine if an immune response is initiated:
A peptide (a protein fragment, possibly from a virus or a mutated cell) must bind to a Human Leukocyte Antigen (HLA) molecule on an antigen-presenting cell.
A T cell Receptor (TCR) on a T cell must then recognize and bind this peptide-HLA (pHLA) complex.
Both binding events are highly specific. This specificity is like a sophisticated lock-and-key system. The complexity arises from the vast diversity of HLA alleles in humans and the even more astounding variety of TCRs. Understanding these binding specificities is crucial for designing effective vaccines and immunotherapies. If we can predict which peptides will strongly bind both HLA and TCR, we can better identify truly immunogenic antigens.
UnifyImmun: A Unified AI Approach for a Two-Part Problem
Previous computational methods often treated pHLA binding and peptide-TCR (pTCR) binding as separate issues. However, an antigen's true immunogenicity depends on both interactions. The creators of UnifyImmun recognized this. They developed a unified cross-attention transformer model to simultaneously predict peptide binding to both HLA and TCR molecules. This integrated method aims for a more holistic view of an antigen's ability to trigger an immune response.
The Architecture: How UnifyImmun Learns the Bind
UnifyImmun has several key components:
Sequence Embedding Blocks: Peptide, HLA, and TCR amino acid sequences are converted into numerical representations (embeddings).
Self-Attention Encoders: Three separate encoders process these embeddings. Self-attention allows the model to weigh the importance of different amino acids within each sequence for feature extraction.
Cross-Attention Layers: These layers integrate features. One fuses peptide and HLA embeddings; another fuses peptide and TCR embeddings. The attention scores from these layers also help pinpoint amino acid sites critical for binding.
Prediction Heads: Outputs from the cross-attention layers go to fully connected layers to predict pHLA and pTCR binding probabilities.

Smart Training for a Complex Task
Training UnifyImmun is challenging, mainly due to the scarcity of experimentally validated HLA-peptide-TCR triplet data. The researchers used clever strategies:
Two-Phase Progressive Training:
Phase 1: The model trains on more abundant pHLA pairwise binding data, keeping TCR-related parts fixed.
Phase 2: It then trains on pTCR pairwise data, with HLA-related parts fixed.
This alternating process, with continuous antigen encoder updates, allows the tasks to mutually reinforce each other. Encoders learn more expressive features. Performance demonstrably improved with more rounds of this strategy.
Virtual Adversarial Training (VAT): To prevent overfitting and improve generalization to unseen data, VAT was employed. This technique introduces small "adversarial" perturbations to sequence embeddings during training. It forces the model to be robust to slight input variations, significantly boosting performance.

Putting UnifyImmun to the Test: Benchmarks and Clinical Relevance
UnifyImmun delivered impressive results:
Superior Predictive Performance:
For pHLA binding, it surpassed 12 other methods on independent and external test sets, with significant AUROC and AUPR improvements (e.g., at least 5% better than TransPHLA on one set ).
For pTCR binding, it clearly outperformed four state-of-the-art methods. On one dataset, UnifyImmun hit AUROC/AUPR of 0.938/0.936; the next best (ERGO2) scored around 0.704/0.747. Others were near random chance.
Shining on a COVID-19 Challenge: On a large COVID-19 pTCR binding test set (over 540,000 interactions, no peptides seen in training), UnifyImmun beat competitors by over 10% in AUROC. This highlights its strong generalisation.
Clinical Correlations: The binding scores predicted by UnifyImmun significantly correlated with patient responses to immunotherapy and clinical outcomes in two melanoma cohorts. Patients with better responses often had higher predicted binding scores for their neoantigens.
Interpretability Pays Off: Cross-attention scores and integrated gradients successfully identified critical amino acid sites for peptide binding. Leu (L) often emerged as important, particularly at the P2 position for both HLA and TCR binding. These findings matched known binding motifs.
My Perspective:
UnifyImmun is a smart step forward in predicting antigen immunogenicity. Its unified framework is more biologically realistic than studying pHLA and pTCR binding separately. The two-phase training and VAT demonstrate clever engineering to maximize performance with limited triplet data and ensure the model generalizes well.
The >10% performance jump on the COVID-19 dataset is a good result. It shows a substantial improvement in handling novel peptides, not just an incremental one. The clinical correlation is key – linking computational predictions to patient immunotherapy outcomes is vital for translational bioinformatics.
The authors acknowledge limitations. The immune response involves more than these two binding events. Also, current TCR repertoire data is limited. However, by openly sharing their model and data (via GitHub and Zenodo ), they enable community contributions. As more TCR data becomes available from single-cell sequencing, and with potential integration of large language model features, models like UnifyImmun will only improve. This work excellently illustrates how sophisticated AI can help decipher one of biology's most complex systems.
Section 3: RNA Takes Center Stage – AI Supercharges the Quest for RNA-Targeting Drugs
The Emerging Frontier: RNA as a Druggable Target
For many years, proteins have dominated drug discovery efforts. Yet, a vast, relatively unexplored landscape exists within our cells: RNA. We now understand that only a small portion of our RNA actually codes for proteins. The remainder (ncRNA) plays crucial roles in numerous biological processes. Imagine the therapeutic possibilities if we could precisely target these RNA molecules with small-molecule drugs. This could unlock new treatments for cancers where protein targets are scarce, or for conditions like triple-negative breast cancer with limited options. The recent FDA approval of the first RNA-targeting drug signals a new era.
Finding drugs that effectively bind to RNA is challenging. Traditional methods like molecular docking simulate how a molecule might fit into an RNA's 3D pocket. However, these are slow and computationally intensive. Screening millions of potential drug compounds this way is a monumental task. Furthermore, there's a data shortage: far fewer RNA-ligand structures and binding affinities are known compared to proteins.

RNAmigos2: Teaching an AI to Spot RNA-Ligand Matches at Lightning Speed
RNAmigos2 is a deep learning pipeline designed to dramatically accelerate structure-based virtual screening for RNA targets. Instead of slow docking simulations, RNAmigos2 learns to predict binding likelihood directly from RNA structure and ligand properties. It achieves an astounding 10,000-fold speedup over traditional docking methods.
The Blueprint: Graphs, Learning, and Smart Ensembles
RNAmigos2 uses a sophisticated pipeline to tackle data scarcity and computational hurdles:
Representing RNA as "2.5D Graphs": Full 3D RNA structures are intricate. RNAmigos2 simplifies them into "2.5D graphs." Nucleotides are nodes, and edges represent both canonical (e.g., Watson-Crick) and non-canonical base-pairing interactions. This captures key 3D architectural features in a discrete format ideal for graph neural networks. Directed edges are used to preserve asymmetries.
Encoding RNA and Ligands:
RNA Encoder: A Relational Graph Convolutional Network (rGCN) creates an "embedding" (a numerical fingerprint) of the RNA binding site graph. It's pre-trained using self-supervision on thousands of RNA substructures to learn general RNA structural motifs.
Ligand Encoder: Small molecules (ligands) are also represented as graphs. A Variational Autoencoder (VAE) architecture encodes them, ensuring their embeddings capture meaningful chemical features.
Dual Decoders for Prediction:
Compatibility Decoder (Compat): Trained as a binary classifier, this decoder distinguishes the native ligand of a binding site from decoys. It uses experimentally known RNA-ligand complexes from the PDB. A margin loss enhances pocket specificity.
Affinity Decoder (Aff): To address limited experimental data, RNAmigos2 uses synthetic data. The team performed large-scale docking experiments (500 drug-like compounds on 1740 binding sites). This decoder predicts these simulated docking scores.
Ensemble Power ("Mixed" Model): Predictions from Compat and Aff decoders are combined (by taking the max of their ranks). This "Mixed" model, the core of RNAmigos2, leverages their complementary strengths. It significantly outperforms either decoder alone and even surpasses standard docking accuracy.

Rigorous Testing: From Benchmarks to Real-World Validation
RNAmigos2 is both fast and accurate:
Outperforming the Competition: On a challenging benchmark with structurally distinct test sets, RNAmigos2 ("Mixed" model) achieved a mean AuROC of 0.972. This was significantly better than rDock (0.959) and other tools. It consistently ranked active compounds in the top 2.8%.
Robustness to Imperfect Pockets: Real-world binding site identification can be inexact. RNAmigos2 showed good robustness even with perturbed or slightly shifted input binding site information. This suggests it can identify the true site from approximate locations.
Experimental Validation – The Clincher: The team tested RNAmigos2 on an independent, large-scale in vitro screen (ROBINS dataset). This involved ~20,000 compounds against unseen RNA riboswitch targets (TPP, ZTP, SAM_II, PreQ1).
RNAmigos2 achieved positive enrichment across all four targets. It significantly distinguished actives from decoys in just two CPU minutes (rDock took ~1000 core hours).
The mean enrichment factor at 1% was an impressive 2.93. This is a landmark: the first experimentally validated success for structure-based deep learning in RNA virtual screening.
The "Aff" model tended to find more generic RNA binders. The "Compat" model, trained on specific PDB complexes, found more pocket-specific binders. RNAmigos2, by combining them, retrieved a more chemically diverse set of active compounds than docking alone.
A Personal Take: The Future is Fast and Graph-Powered!
RNAmigos2 is a genuinely exciting development for anyone at the crossroads of computation and biology. The 10,000x speed-up is not just an incremental gain; it changes the landscape of what's achievable in RNA drug discovery. We can now feasibly screen massive compound libraries.
Using 2.5D graphs is a clever method to balance structural detail with computational efficiency. The dual-decoder approach, especially with synthetic docking data, smartly navigates the current scarcity of experimental RNA-ligand data. The fact that this model performs well both in silico and in a large experimental screen is compelling validation.
Limitations exist, of course. A predefined binding site is necessary. Modelling RNA flexibility also remains a significant hurdle. However, the authors are already looking forward. They suggest integrating with binding site predictors and exploring other docking tools for even more diverse surrogate models. With tools like AlphaFold3 now also predicting RNA structures, the potential synergy is clear. RNAmigos2 is a powerful new asset, and I eagerly anticipate the discoveries it will facilitate.
Conclusion: The Algorithmic Renaissance in Biology
The three studies explored today—PSAM illuminating cancer mutations, UnifyImmun decoding immune dialogues, and RNAmigos2 accelerating RNA drug discovery —are vibrant illustrations of deep learning's impact on biomedicine. Each model, with its unique design and intelligent training, pushes the limits of our predictive and analytical capabilities in biological systems.
Key Themes We've Seen:
Beyond Human Intuition: Deep learning models uncover subtle patterns and non-obvious relationships in biological data that might elude traditional methods.
The Power of Integration: Combining different neural network types (CNNs, BiLSTMs, GNNs), attention mechanisms, and multi-task learning leads to more robust and insightful models.
Bridging Computation and Experiment: The most compelling AI tools are rigorously validated against real-world experimental data and clinical outcomes. This prediction-validation-refinement cycle is essential.
Interpretability Matters: While "black box" models can be potent, the move towards more interpretable AI (using attention scores or residue-level contributions) allows us to understand why a model makes its predictions. This, in turn, can generate new biological hypotheses.
As we stand at this exciting juncture, the fusion of sophisticated algorithms with deep biological knowledge is clearly the path forward. The ability to predict and understand health and disease mechanisms at such a detailed level promises to revolutionize diagnostics, therapy design, and potentially, disease prevention. The journey is complex, and challenges remain, but the potential for discovery is immense.
Thank you for joining this deep dive. What are your thoughts on these advancements? Where else do you see AI making significant contributions in biology? Let's discuss!
Sources
Zheng, Y., Yu, K., Lin, JF. et al. Deep learning prioritizes cancer mutations that alter protein nucleocytoplasmic shuttling to drive tumorigenesis. Nat Commun 16, 2511 (2025). https://doi.org/10.1038/s41467-025-57858-8
Yu, C., Fang, X., Tian, S. et al. A unified cross-attention model for predicting antigen binding specificity to both HLA and TCR molecules. Nat Mach Intell 7, 278–292 (2025). https://doi.org/10.1038/s42256-024-00973-w
Carvajal-Patiño, J.G., Mallet, V., Becerra, D. et al. RNAmigos2: accelerated structure-based RNA virtual screening with deep graph learning. Nat Commun 16, 2799 (2025). https://doi.org/10.1038/s41467-025-57852-0
Excellent article! Wish all the best for you and your family.
Wishing you well!