


On New cells in the Brain and Blood.
How are the researchers leveraging single-cell technologies to discover new cell types and find the evolutionary conservation across organs?

There are a plethora of challenges to identify and illuminate the broad sets of cells present in various organisms across the tree of life. Historically, pathologists and cell biologists have relied on morphology, function and tissue of origin to classify cells and their roles in the organism. But morphological classification has its limitations, as you can imagine there are more cells that might look similar and function very differently. The introduction of sensitive methods to identify the surface proteins and assigning functions based on these observations either using knockdown or knockout experiments eventually superseded the old observation under the microscope and report it with more robust classifications of cells. In a similar fashion, the introduction of scSEQ and further developments in platforms such as 10x Genomics and slide-SEQ, has now allowed scientist to dig deeper into types of cells and gene expression patterns in single cells giving us more meaningful insights into the complex interplay between gene expression and cell type identification based on known or novel gene markers.
This week, There have been two papers that grabbed my attention in regard to the cell type problem. A study led by Ruth Styfhals and colleagues asked for a quirky and very important question “What do octopuses, mice, and the fruit fly share when it comes to their brains and the cells of their brains” and Rebecca Boiarsky and colleagues ask “Can we find aberrant gene expression alterations in Multiple Myeloma?”
Lets take the papers one by one and explore some of the key questions addressed and how they did that.
Diversity of cell types in octopus brains

Photo by Isabel Galvez on Unsplash
Octopuses are one of the most fascinating creatures for me, From potentially predicting the World cup results to being star of the Netflix documentary “My Octopus Teacher” or just being wickedly smart and outsmarting other reef creatures, these molluscs seem to be able to do perform multi-tasking as it is in their second nature. With a large centralised brain with more than 30 different lobes performing computations, transfers and integrations of information they indeed have a complex brain amongst the invertebrates, approximately 200 million cells work together to keep this reef predator going. Work published by John Zachary young (this book, and the article) describe the morphological characteristics. But, we still did not know what are the functions of different cells that are present.
The authors used the paralarval stage of the life cycle of the octopus as a starting point as it contains approximately 200,000 cells to build an atlas. The atlas aims to systematically characterise and compare across species to find the cell-type diversity.
What did they do?
From a one-day-old Octopus vulgaris, the authors performed both single cell and single nuclei sequencing and used few other methods such as FLAM-Seq and Iso-Seq to improve the poorly annotated transcriptome. A total of 8564 cells and 8517 nuclei were used to perform further analysis. One might ask, Why do you need nuclei and cells? By integrating both of them, the authors aimed to get the most information about the cell types and reduce any artefacts that one method produces.






What are the initial observations?
Cells seem to produce transcripts associated to immediate early genes and heat shock proteins.
The early growth response gene (egr1) was switched on.
They identified previously known cell types such as dopaminergic, GABAergic, serotonergic and peptidergic neurons. Also, 3 different FMRFamide precursor genes seem to be expressed differently between cell types.
Some types of neurons which were previously thought to be only in adults were already seen at a hatching stage.
Critical differences in cell states was reported between the periphery and central constellation, these differences were partly attributed to transcription factors like dimm & rn/sqz (central constellation) tfam & bcl11a/b (other surrounding clusters).
Cells of the central constellation were transcriptionally diverse indicating activity
t-SNE was similar to what was observed in Drosophilia which was mapped to the central brain of the octopus.
The central constellation is a heterogenous cluster of cells transcriptionally similar to the optic lobe or central brain and contains rare cell types.
Within the retina: They found 3 distinct cell types within the outer granular layer of the optic lobe.
While cross-species cell type comparisons using SAMap algorithm they found few similarities to glia and astrocytes in fly and mouse respectively. They could also deduce that octopus-fly-mouse glial cells share conserved gene expression with orthologous genes.
Structure-wise: the authors were curious about these cell similarities and abundant differences so they tried to identify genes that usually discriminate with fly glial subtypes. They show that there are significant differences; of the three distinct glial subtypes identified
GLIA1 mainly expressed GABA transporter 1, by using two other markers they show that some cells in GLIA1 strongly resemble astrocytes.
Bassed where in the brain GLIA1 cells reside they were able to distinguish between the neuropil glia and infiltrating glia which likely provides support and neuromodulation like vertebrates(Still speculation).
GLIA2 seems to differentially express neurotransmitters and adhesion molecules.
GLIA3 seems to express vascular endothelial growth factor and very few genes involved in neuromodulation
“We, therefore, hypothesize that GLIA2 are the glia in the plexiform layer and that GLIA3 form a membrane surrounding the brain (not directly in contact with the neurons), and might play a role in the hemolymphbrain-barrier, similar to astrocytes. Both GLIA1 and GLIA3 mapped to mouse astrocytes, while GLIA2 was not identified in the cross-species mappings”
Using SAMap while comparing Vertical lobe and fly gamma kenyon cells since Vl is considered to be centre for learning and memory in octopus.
VL marker genes aristaless, pka-R2 and tmtc4 were expressed and with vacht suggesting that these cells are cholinergic amacrine cells in adult VL.
These cells are involved in long-term memory potentiation and formation.
Expressed genes found in “mushroom-body” in the fly were also enriched within the VL.
This may indicate that gene expression profiles either have a common origin or common functions.
“These findings suggest that some cell types might deploy deeply conserved transcriptional programs across bilaterian evolution”
What exactly makes these cells different from each other?
Transcription factors are known molecules that dictate cell fate during development.
Top differentially expressed TF’s are shown below in a tree form below

Most dopaminergic celltypes expressed a ZnF TF gene: branch h above
Branch D, are cells which are all localized in the (sub)vertical lobe
Branches D-FL central brain cell types and Branches G-K are optic lobe cell types.
Homeobox transcription factors and their combinations there of seem to determine the cell fates in octopus developing brains.
In summary:
42 of the possible and likely 116 cell types of adult octopus brains were described in the paper to be seen/expressed in developing hatching brains.
A large number of cells in the central constellation likely are rare cell types.
Transcription factors of the homeobox family and ZnF family seem to dictate cell fate.
One of the first attempts to understand cell type diversity and complexity in octopuses
enables comparative study across several diverse range of invertebrates.
Cell atlas-based approach and comparative genomics are interesting approaches to understanding the less known organisms with which we share the world.
Of the rare, but even more uncommon
Multiple myeloma (MM) is are relatively uncommon cancer with the life time risk of getting MM in the US is 0.76%. As the name suggests the disease pertains to the myeloid cells especially the plasma cells that are usually found in the bone marrow. Plasma cells are heavily involved in fight against pathogens since they are involved in production of antibodies which are transported from plasma cells by the blood plasma (learn about cell lineages). The interesting thing about MM is that, as a disease it has always shown some pre-cursor states of the malignant cell but with a highly heterogenous outcome. There is an urgent need to define the molecular characteristics of patients who are at risk of progression.
What did they do?
The authors in this work were interested in identifying few basic questions regarding MM. Since it is known that MM is preceded by precursor stage knowns as MGUS and SMM, can we find subpopulation of cells that have distinct changes so that the disease progression from a relatively mild MGUS to severe MM can be identified early?
Single-cell RNA-sequencing of plasma cells from 26 patients at varying stages of the disease and 9 healthy samples as controls to compare the data.
29,387 plasma cells a total representation of the samples were profiled.
They use an Automatic relevance determination non-negative matrix factorization (ARF-NMF) was created and used to highlight gene signatures that were active in their cohort and validated with external cohorts.
Taken together, our study (i) presents a highly detailed and comprehensive view of the transcriptional transformation occurring in individual patients with myeloma and its precursor conditions, (ii) discovers gene expression signatures that are shared across patients with different driver events and at different stages of disease, and (iii) characterizes heterogeneity both between and within tumors
What are their observations?
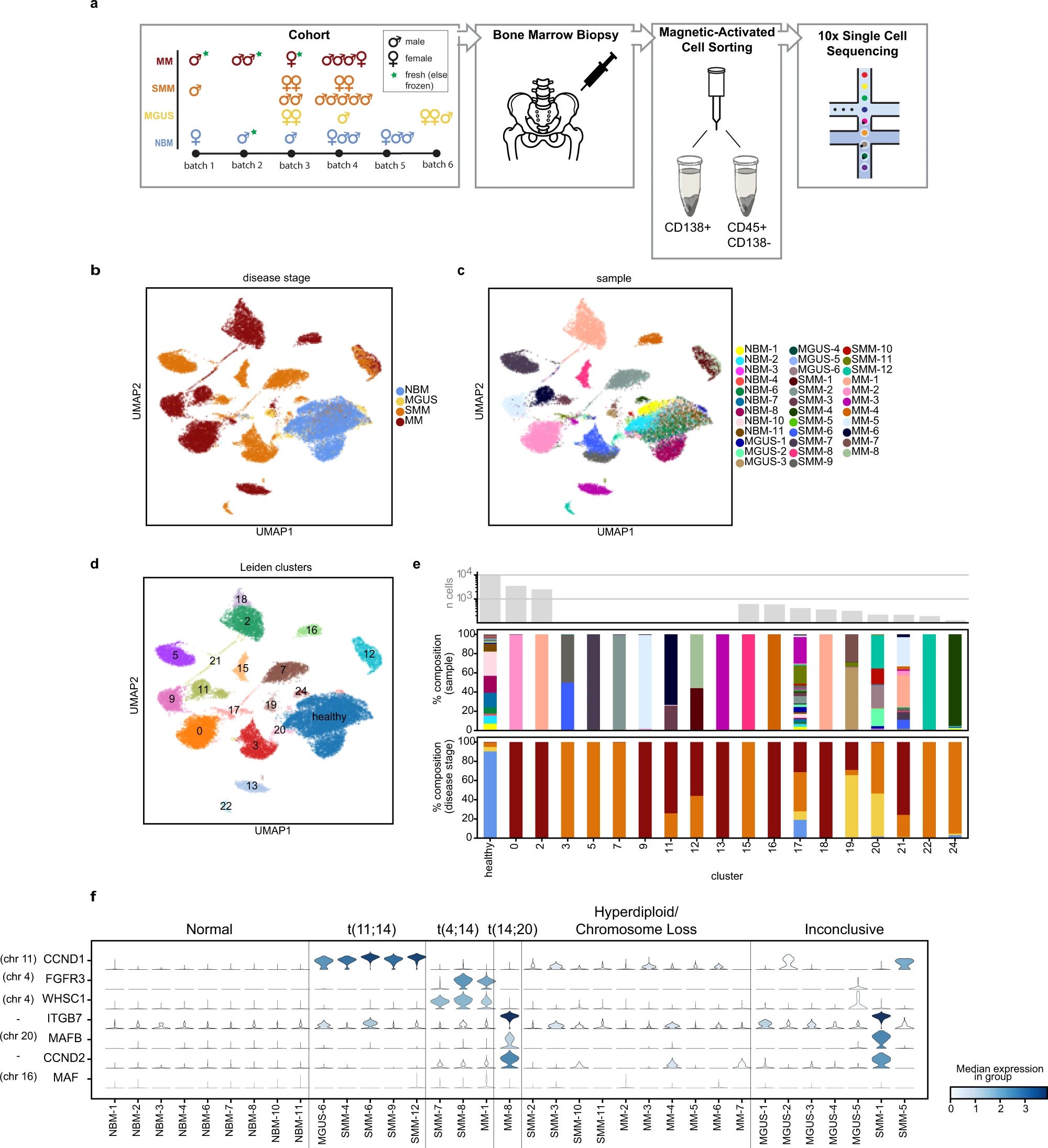
Using clustering they obtained 25 clusters of cells. 7 represented healthy cells, 11 clusters were single sample origin only, and the rest were identified on the basis of the patient and abnormally proliferating cells across the disease stages.
Observations suggest that the patients samples on displayed a variety of cell normal plasma to abnormal plasma cells (MGUS had 73% normal vs 8% in SMM)
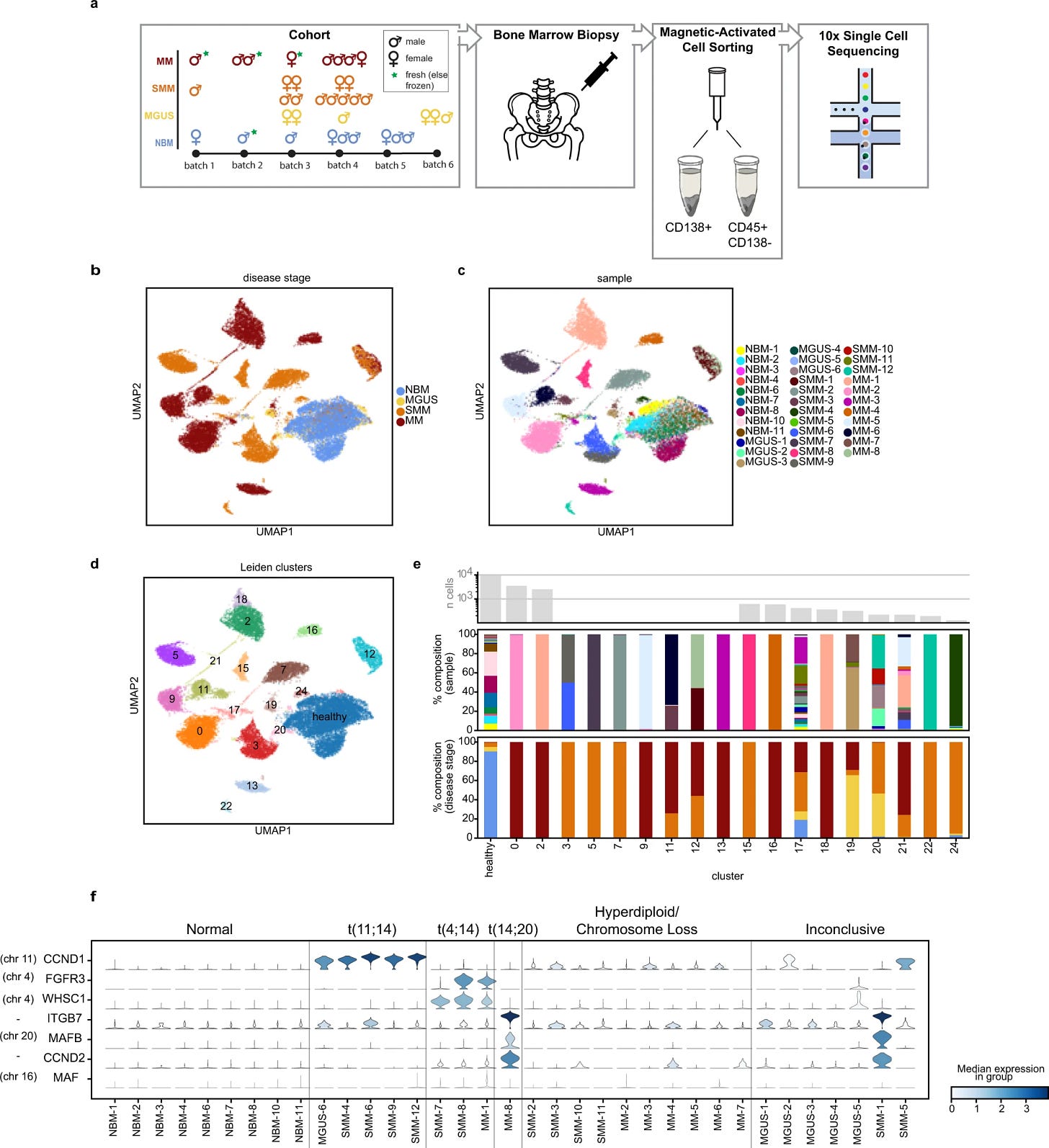
Figures showing the experimental set-up and clustering of the cells based on disease stage, sample label and integrated clusters(a-d). E shows us a nice representation of sample composition based on clustering in E. F is finally the distribution of expression of genes commonly upregulated. by pseudosampling (considering all normal cells vs abnormal cells) they were able to identify 764 Differentially expressed genes
Up regulated: RBFOX2, STIM1, IFIT1, Rp11-395G23.3
Down regulated CD19, CTSH, CD81, ITGB2

Certain pathways such as interferon alpha response and Wnt/ß-catenin signalling were differentially enriched. Overall providing a general view of genes whose expression is consistently altered in disease.
To address the inter-patient heterogeneity and patient-specific disease characteristics
They compared the gene expression of abnormal plasma cells vs the patient-derived healthy cell population. This gives rise to unique profiles of individual tumors.
This partitioning of the sample led to identifying 1760 DEGs and in conjunction with 764 DEGS we see an overlap of 251 DEGs
Interesting genes include GNB2L1, HIST1H1C, CD59 and MYC



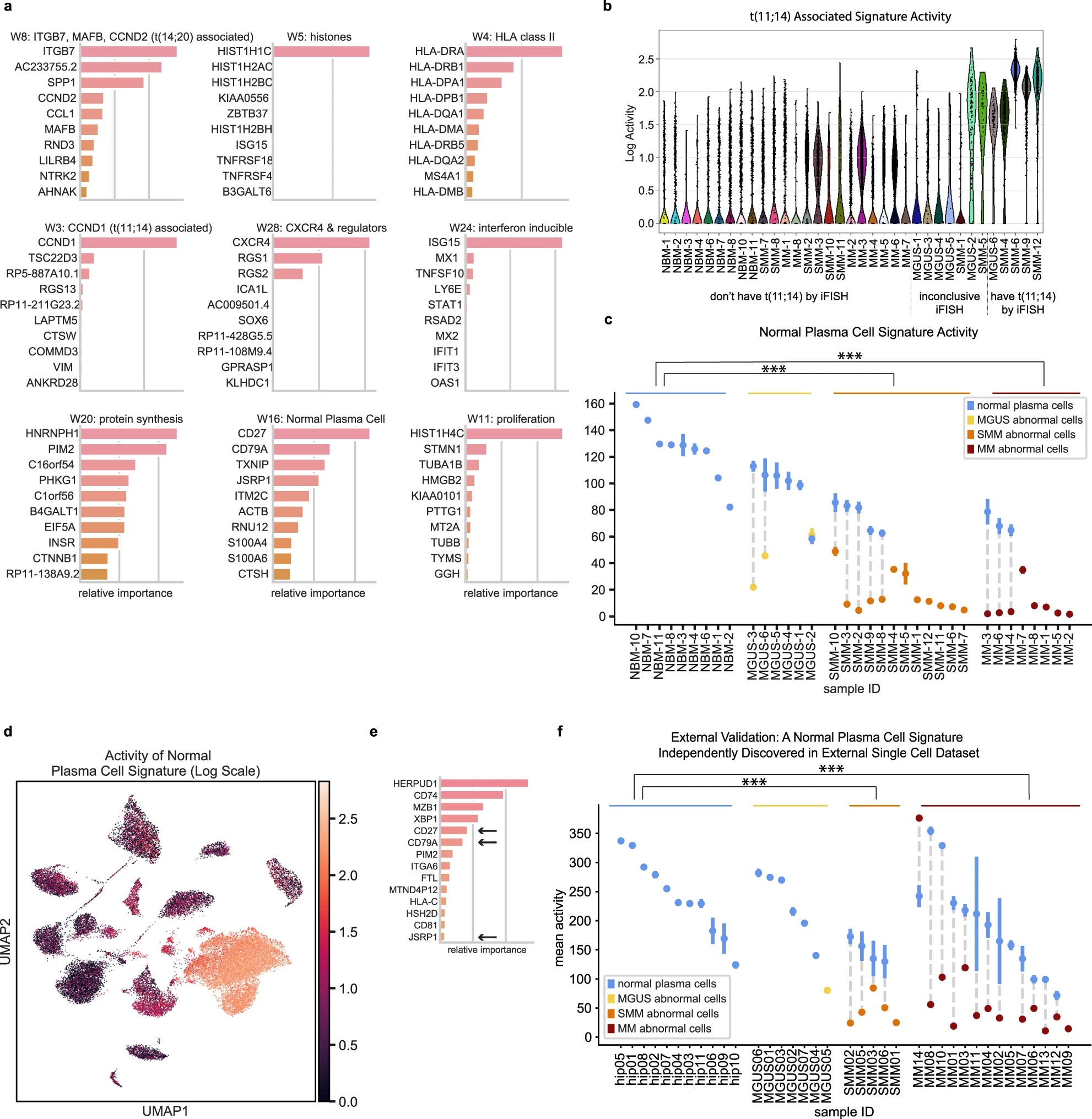
ARD-NMF: The automatic relevance determination NMF is based on a work by VYF Tan and C Févotte. It essentially is a matrix multiplication method where the gene expression profile of each cell is an additive combination of latent gene expression signature and associated weights. Learn more about NMF here
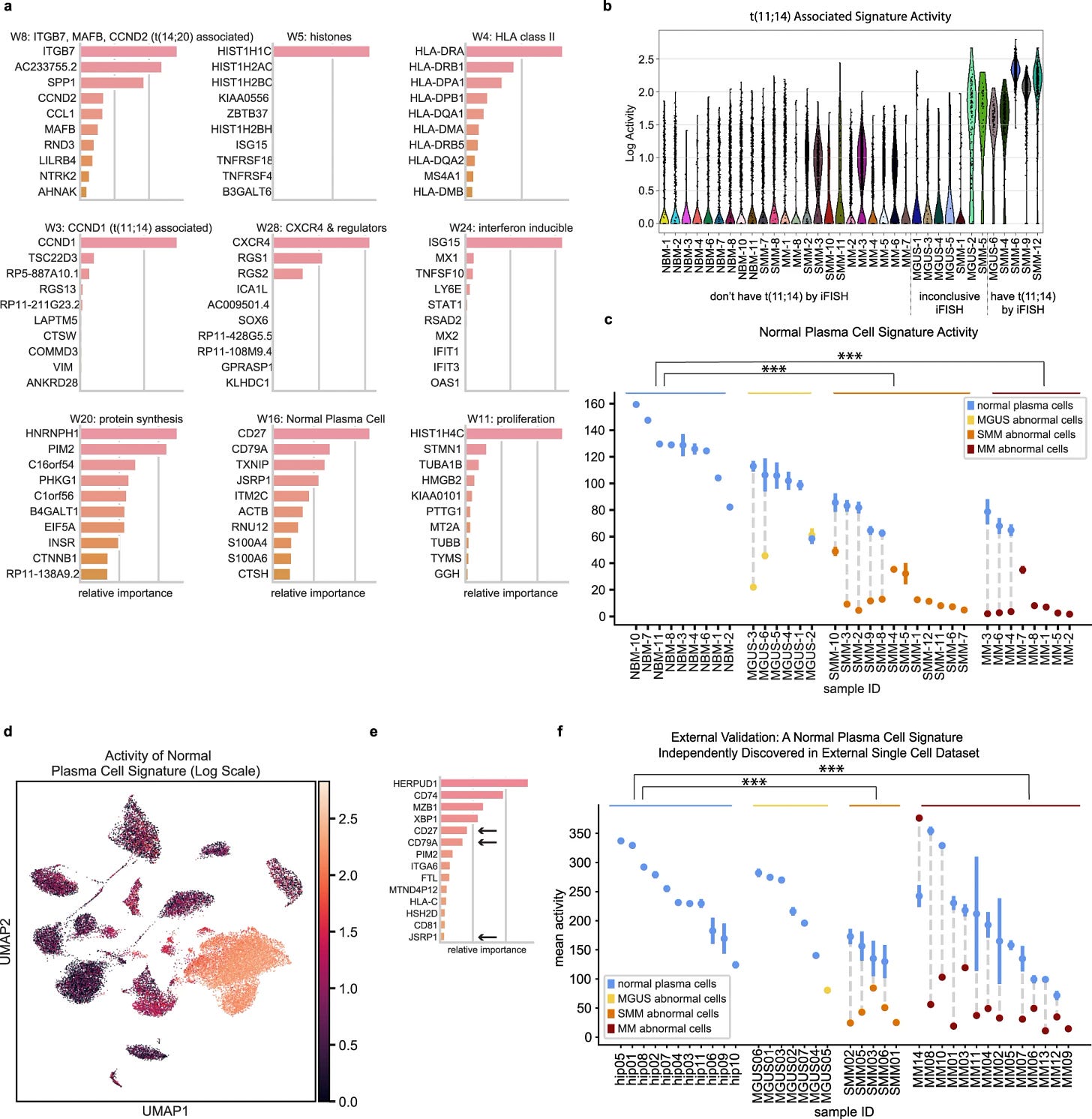
NMF based data interpretation to identify sensitive signatures across myeloma cells Using the method they generated 28 gene signatures.
Removed signatures that were active for single patients. This left 15 signatures, 9 of them are shown in the figure with further investigations.

15 of the gene expression signatures discovered using ARD-NMF
They discovered that ’normal plasma cell signature’ is down regulated in abnormal cells with CD27, CD79A as well as JSRP1, CTHS HCST and RNU12
They identified that the scSEQ method can identify a low abundance signature that seems to be not present in bulk RNA-seq and the process of signature changes occurs as early as MGUS stage.
They validated the findings with single-cell data from Ledergor et al and recovered similar signatures. Supporting that these signatures can be potentially used for early detection of tumour progression.
They also found that patients with high IFN-inducible gene signature activity in their T cells also had a high activity of the IFN signature in their CD138+ cells.

In summary:
They identified a signature present in normal plasma cells but uniformly lost at all stages of MM progression. CD27, one of the top genes of this signature, has been previously discussed in MM literature but has been reported to have a variable expression in myeloma cells, increased expression in MGUS, and a correlation with prognosis.
abnormal plasma cells had lower expression of another mature B cell marker, CD79A, as well as decreased enrichment of immune pathways, such as the complement pathway.
Pathway level transcriptional changes: aberrant expression of Wnt pathway members including overexpression of DKK1 in abnormal cells of precursor myeloma. Given that many cases of MGUS also have osteoporosis and osteopenia, this may indicate that Wnt dysregulation and DKK1 overexpression are associated with early osteopenia in those patients and are potentially predictors of the development of osteolytic lesions
Interferon signaling in myeloma cells, which has been reported previously53, may be a response to a common stimulus in the microenvironment that affects multiple cell types, including normal plasma cell.
Within-patient DE analysis points to potential therapeutic targets that are present in subsets of patients not only in MM, but also in earlier disease stages, and which can be selected for functional validation.
Takeaways
scSEQ is a fascinating new tool to discover new and novel cell types or to identify rare transitionary states in diseases
As a bioinformatician, One can see unique sets of challenges like the absence of a good reference genome for octopus or how to apply NMF for extracting common/unique modalities.
Also, How cool is it if we were able to make SC-atlases of all living creatures?
LINKS:
Ruth’s tweet on the paper: https://tinyurl.com/3jc255r9 & please follow Ruth, for further research that she might be a part of.
The Seuntjens Lab : https://bio.kuleuven.be/df/es/
The full-text article: https://tinyurl.com/23b3x725
The Getz lab tweet on the paper: https://tinyurl.com/5xa3fyc7, https://tinyurl.com/2p99p53n, follow Rebecca here
The Getz lab: https://www.broadinstitute.org/labs/getz
The full-text article: https://tinyurl.com/yemdspvx
Do you like the article? And do you want a place to hang out, talk, and be the nerd you are meant to be? Subscribe now for free and spread the word!!!
oh, If by you gained some insights and would like to help me be caffeinated to write the posts please scan the QR below or click here :)
